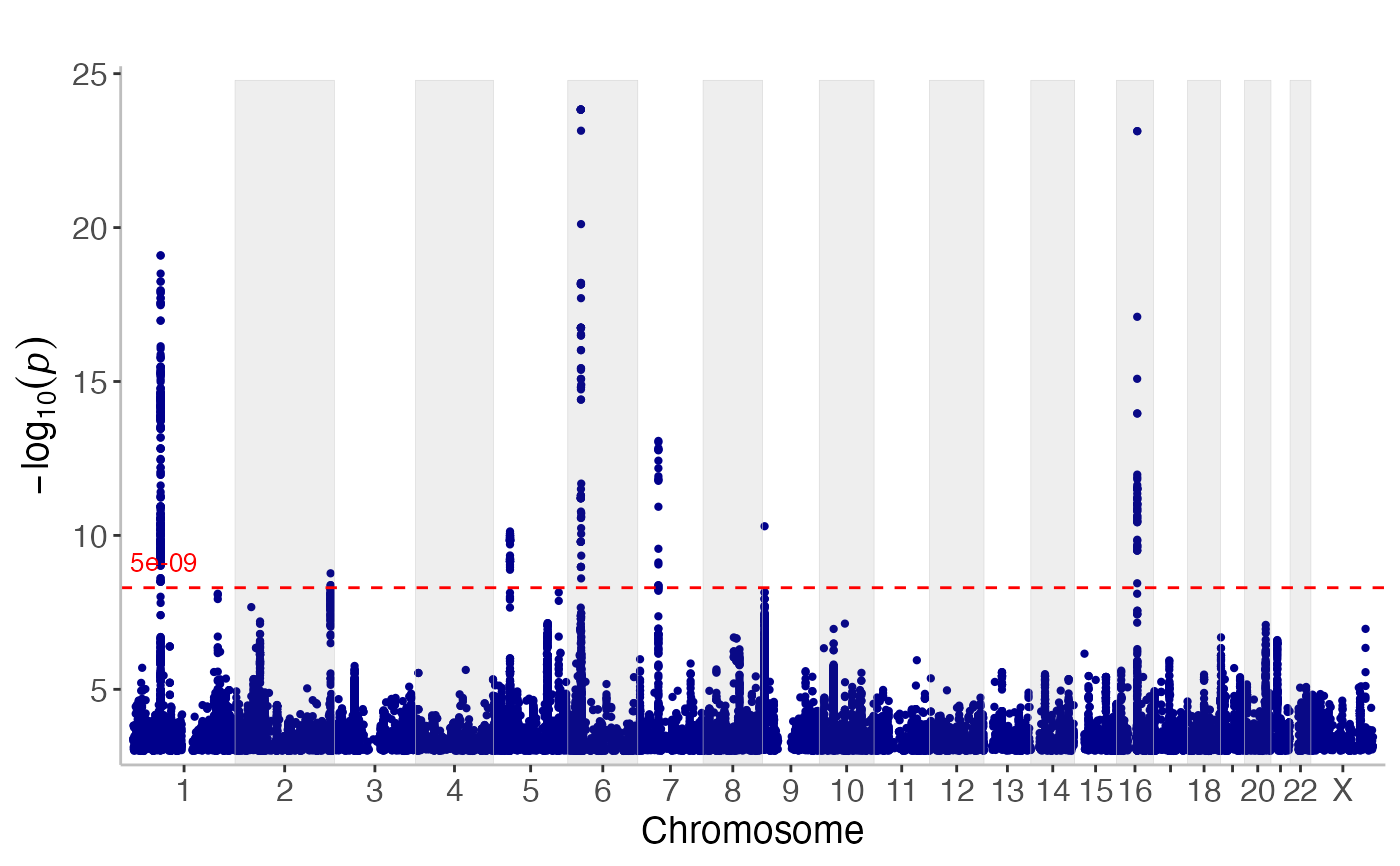
manhattan() displays association results for the entire genome on a Manhattan plot.
Required parameter is at least one dataset (dataframe) containing the association data (with columns CHROM,POS,P in upper or lowercase)
All other input parameters are optional
manhattan(
df,
ntop = 4,
title = "",
annotate = NULL,
color = get_topr_colors(),
sign_thresh = 5e-09,
sign_thresh_color = "red",
sign_thresh_label_size = 3.5,
label_size = 3.5,
size = 0.8,
shape = 19,
alpha = 1,
highlight_genes_color = "green",
highlight_genes_ypos = 1,
axis_text_size = 12,
axis_title_size = 14,
title_text_size = 15,
legend_title_size = 13,
legend_text_size = 12,
protein_coding_only = TRUE,
angle = 0,
legend_labels = NULL,
chr = NULL,
annotate_with = "Gene_Symbol",
region_size = 1e+06,
legend_name = NULL,
legend_position = "bottom",
nudge_x = 0.1,
nudge_y = 0.2,
xmin = NULL,
xmax = NULL,
ymin = NULL,
ymax = NULL,
highlight_genes = NULL,
label_color = NULL,
legend_nrow = NULL,
gene_label_size = NULL,
gene_label_angle = 0,
scale = 1,
show_legend = TRUE,
sign_thresh_linetype = "dashed",
sign_thresh_size = 0.5,
rsids = NULL,
rsids_color = NULL,
rsids_with_vline = NULL,
annotate_with_vline = NULL,
shades_color = NULL,
shades_alpha = 0.1,
segment.size = 0.2,
segment.color = "black",
segment.linetype = "solid",
max.overlaps = 10,
label_fontface = "plain",
label_family = "",
gene_label_fontface = "plain",
gene_label_family = "",
build = 38,
verbose = NULL,
label_alpha = 1,
shades_line_alpha = 1
)Arguments
- df
Dataframe or a list of dataframes (required columns are
CHROM,POS,P), in upper- or lowercase) of association results.- ntop
An integer, number of datasets (GWASes) to show on the top plot
- title
A string to set the plot title
- annotate
A number (p-value). Display annotation for variants with p-values below this threshold
- color
A string or a vector of strings, for setting the color of the datapoints on the plot
- sign_thresh
A number or vector of numbers, setting the horizontal significance threshold (default:
sign_thresh=5.1e-9). Set to NULL to hide the significance threshold.- sign_thresh_color
A string or vector of strings to set the color/s of the significance threshold/s
- sign_thresh_label_size
A number setting the text size of the label for the significance thresholds (default text size is 3.5)
- label_size
An number to set the size of the plot labels (default:
label_size=3)- size
A number or a vector of numbers, setting the size of the plot points (default:
size=1.2)- shape
A number of a vector of numbers setting the shape of the plotted points
- alpha
A number or a vector of numbers setting the transparency of the plotted points
- highlight_genes_color
A string, color for the highlighted genes (default: green)
- highlight_genes_ypos
An integer, controlling where on the y-axis the highlighted genes are placed (default value is 1)
- axis_text_size
A number, size of the x and y axes tick labels (default: 12)
- axis_title_size
A number, size of the x and y title labels (default: 12)
- title_text_size
A number, size of the plot title (default: 13)
- legend_title_size
A number, size of the legend title
- legend_text_size
A number, size of the legend text
- protein_coding_only
A logical scalar, if TRUE, only protein coding genes are used for annotation
- angle
A number, the angle of the text label
- legend_labels
A string or vector of strings representing legend labels for the input dataset's
- chr
A string or integer, the chromosome to plot (i.e. chr15), only required if the input dataframe contains results from more than one chromosome
- annotate_with
A string. Annotate the variants with either Gene_Symbol or ID (default: "Gene_Symbol")
- region_size
An integer (default = 1000000) (or a string represented as 100kb or 1MB) indicating the window size for variant labeling. Increase this number for sparser annotation and decrease for denser annotation.
- legend_name
A string, use to change the name of the legend (default: None)
- legend_position
A string, top,bottom,left or right
- nudge_x
A number to vertically adjust the starting position of each gene label (this is a ggrepel parameter)
- nudge_y
A number to horizontally adjust the starting position of each gene label (this is a ggrepel parameter)
- xmin, xmax
Integer, setting the chromosomal range to display on the x-axis
- ymin, ymax
Integer, min and max of the y-axis, (default values:
ymin=0, ymax=max(-log10(df$P)))- highlight_genes
A string or vector of strings, gene or genes to highlight at the bottom of the plot
- label_color
A string or a vector of strings. To change the color of the gene or variant labels
- legend_nrow
An integer, sets the number of rows allowed for the legend labels
- gene_label_size
A number setting the size of the gene labels shown at the bottom of the plot
- gene_label_angle
A number setting the angle of the gene label shown at the bottom of the plot (default: 0)
- scale
A number, to change the size of the title and axes labels and ticks at the same time (default = 1)
- show_legend
A logical scalar, set to FALSE to hide the legend (default value is TRUE)
- sign_thresh_linetype
A string, the line-type of the horizontal significance threshold (default = dashed)
- sign_thresh_size
A number, sets the size of the horizontal significance threshold line (default = 1)
- rsids
A string (rsid) or vector of strings to highlight on the plot, e.g.
rsids=c("rs1234, rs45898")- rsids_color
A string, the color of the variants in variants_id (default color is red)
- rsids_with_vline
A string (rsid) or vector of strings to highlight on the plot with their rsids and vertical lines further highlighting their positions
- annotate_with_vline
A number (p-value). Display annotation and vertical lines for variants with p-values below this threshold
- shades_color
The color of the rectangles (shades) representing the different chromosomes on the Manhattan plot
- shades_alpha
The transparency (alpha) of the rectangles (shades)
- segment.size
line segment color (ggrepel argument)
- segment.color
line segment thickness (ggrepel argument)
- segment.linetype
line segment solid, dashed, etc.(ggrepel argument)
- max.overlaps
Exclude text labels that overlap too many things. Defaults to 10 (ggrepel argument)
- label_fontface
A string or a vector of strings. Label font “plain”, “bold”, “italic”, “bold.italic” (ggrepel argument)
- label_family
A stirng or a vector of strings. Label font name (default ggrepel argument is "")
- gene_label_fontface
Gene label font “plain”, “bold”, “italic”, “bold.italic” (ggrepel argument)
- gene_label_family
Gene label font name (default ggrepel argument is "")
- build
A number representing the genome build. Set to 37 to change to build (GRCh37). The default is build 38 (GRCh38).
- verbose
A logical scalar (default: NULL). Set to FALSE to suppress printed messages
- label_alpha
An number or vector of numbers to set the transparency of the plot labels (default:
label_alpha=1)- shades_line_alpha
The transparency (alpha) of the lines around the rectangles (shades)
Value
ggplot object
Examples
manhattan(CD_UKBB)